Spaces:
Sleeping

Sleeping
| import os,sys | |
| # install environment goods | |
| #os.system("pip -q install dgl -f https://data.dgl.ai/wheels/cu113/repo.html") | |
| os.system('pip install dgl==1.0.2+cu116 -f https://data.dgl.ai/wheels/cu116/repo.html') | |
| #os.system('pip install gradio') | |
| os.environ["DGLBACKEND"] = "pytorch" | |
| #os.system(f'pip install -r ./PROTEIN_GENERATOR/requirements.txt') | |
| print('Modules installed') | |
| os.system('pip install --force gradio==3.28.3') | |
| os.environ["DGLBACKEND"] = "pytorch" | |
| if not os.path.exists('./SEQDIFF_230205_dssp_hotspots_25mask_EQtasks_mod30.pt'): | |
| print('Downloading model weights 1') | |
| os.system('wget http://files.ipd.uw.edu/pub/sequence_diffusion/checkpoints/SEQDIFF_230205_dssp_hotspots_25mask_EQtasks_mod30.pt') | |
| print('Successfully Downloaded') | |
| if not os.path.exists('./SEQDIFF_221219_equalTASKS_nostrSELFCOND_mod30.pt'): | |
| print('Downloading model weights 2') | |
| os.system('wget http://files.ipd.uw.edu/pub/sequence_diffusion/checkpoints/SEQDIFF_221219_equalTASKS_nostrSELFCOND_mod30.pt') | |
| print('Successfully Downloaded') | |
| import numpy as np | |
| import gradio as gr | |
| import py3Dmol | |
| from io import StringIO | |
| import json | |
| import secrets | |
| import copy | |
| import matplotlib.pyplot as plt | |
| from utils.sampler import HuggingFace_sampler | |
| plt.rcParams.update({'font.size': 13}) | |
| with open('./tmp/args.json','r') as f: | |
| args = json.load(f) | |
| # manually set checkpoint to load | |
| args['checkpoint'] = None | |
| args['dump_trb'] = False | |
| args['dump_args'] = True | |
| args['save_best_plddt'] = True | |
| args['T'] = 25 | |
| args['strand_bias'] = 0.0 | |
| args['loop_bias'] = 0.0 | |
| args['helix_bias'] = 0.0 | |
| def protein_diffusion_model(sequence, seq_len, helix_bias, strand_bias, loop_bias, | |
| secondary_structure, aa_bias, aa_bias_potential, | |
| #target_charge, target_ph, charge_potential, | |
| num_steps, noise, hydrophobic_target_score, hydrophobic_potential): | |
| dssp_checkpoint = './SEQDIFF_230205_dssp_hotspots_25mask_EQtasks_mod30.pt' | |
| og_checkpoint = './SEQDIFF_221219_equalTASKS_nostrSELFCOND_mod30.pt' | |
| model_args = copy.deepcopy(args) | |
| # make sampler | |
| S = HuggingFace_sampler(args=model_args) | |
| # get random prefix | |
| S.out_prefix = './tmp/'+secrets.token_hex(nbytes=10).upper() | |
| # set args | |
| S.args['checkpoint'] = None | |
| S.args['dump_trb'] = False | |
| S.args['dump_args'] = True | |
| S.args['save_best_plddt'] = True | |
| S.args['T'] = 20 | |
| S.args['strand_bias'] = 0.0 | |
| S.args['loop_bias'] = 0.0 | |
| S.args['helix_bias'] = 0.0 | |
| S.args['potentials'] = None | |
| S.args['potential_scale'] = None | |
| S.args['aa_composition'] = None | |
| # get sequence if entered and make sure all chars are valid | |
| alt_aa_dict = {'B':['D','N'],'J':['I','L'],'U':['C'],'Z':['E','Q'],'O':['K']} | |
| if sequence not in ['',None]: | |
| L = len(sequence) | |
| aa_seq = [] | |
| for aa in sequence.upper(): | |
| if aa in alt_aa_dict.keys(): | |
| aa_seq.append(np.random.choice(alt_aa_dict[aa])) | |
| else: | |
| aa_seq.append(aa) | |
| S.args['sequence'] = aa_seq | |
| else: | |
| S.args['contigs'] = [f'{seq_len}'] | |
| L = int(seq_len) | |
| if secondary_structure in ['',None]: | |
| secondary_structure = None | |
| else: | |
| secondary_structure = ''.join(['E' if x == 'S' else x for x in secondary_structure]) | |
| if L < len(secondary_structure): | |
| secondary_structure = secondary_structure[:len(sequence)] | |
| elif L == len(secondary_structure): | |
| pass | |
| else: | |
| dseq = L - len(secondary_structure) | |
| secondary_structure += secondary_structure[-1]*dseq | |
| # potentials | |
| potential_list = [] | |
| potential_bias_list = [] | |
| if aa_bias not in ['',None]: | |
| potential_list.append('aa_bias') | |
| S.args['aa_composition'] = aa_bias | |
| if aa_bias_potential in ['',None]: | |
| aa_bias_potential = 3 | |
| potential_bias_list.append(str(aa_bias_potential)) | |
| ''' | |
| if target_charge not in ['',None]: | |
| potential_list.append('charge') | |
| if charge_potential in ['',None]: | |
| charge_potential = 1 | |
| potential_bias_list.append(str(charge_potential)) | |
| S.args['target_charge'] = float(target_charge) | |
| if target_ph in ['',None]: | |
| target_ph = 7.4 | |
| S.args['target_pH'] = float(target_ph) | |
| ''' | |
| if hydrophobic_target_score not in ['',None]: | |
| potential_list.append('hydrophobic') | |
| S.args['hydrophobic_score'] = float(hydrophobic_target_score) | |
| if hydrophobic_potential in ['',None]: | |
| hydrophobic_potential = 3 | |
| potential_bias_list.append(str(hydrophobic_potential)) | |
| if len(potential_list) > 0: | |
| S.args['potentials'] = ','.join(potential_list) | |
| S.args['potential_scale'] = ','.join(potential_bias_list) | |
| # normalise secondary_structure bias from range 0-0.3 | |
| S.args['secondary_structure'] = secondary_structure | |
| S.args['helix_bias'] = helix_bias | |
| S.args['strand_bias'] = strand_bias | |
| S.args['loop_bias'] = loop_bias | |
| # set T | |
| if num_steps in ['',None]: | |
| S.args['T'] = 20 | |
| else: | |
| S.args['T'] = int(num_steps) | |
| # noise | |
| if 'normal' in noise: | |
| S.args['sample_distribution'] = noise | |
| S.args['sample_distribution_gmm_means'] = [0] | |
| S.args['sample_distribution_gmm_variances'] = [1] | |
| elif 'gmm2' in noise: | |
| S.args['sample_distribution'] = noise | |
| S.args['sample_distribution_gmm_means'] = [-1,1] | |
| S.args['sample_distribution_gmm_variances'] = [1,1] | |
| elif 'gmm3' in noise: | |
| S.args['sample_distribution'] = noise | |
| S.args['sample_distribution_gmm_means'] = [-1,0,1] | |
| S.args['sample_distribution_gmm_variances'] = [1,1,1] | |
| if secondary_structure not in ['',None] or helix_bias+strand_bias+loop_bias > 0: | |
| S.args['checkpoint'] = dssp_checkpoint | |
| S.args['d_t1d'] = 29 | |
| print('using dssp checkpoint') | |
| else: | |
| S.args['checkpoint'] = og_checkpoint | |
| S.args['d_t1d'] = 24 | |
| print('using og checkpoint') | |
| for k,v in S.args.items(): | |
| print(f"{k} --> {v}") | |
| # init S | |
| S.model_init() | |
| S.diffuser_init() | |
| S.setup() | |
| # sampling loop | |
| plddt_data = [] | |
| for j in range(S.max_t): | |
| output_seq, output_pdb, plddt = S.take_step_get_outputs(j) | |
| plddt_data.append(plddt) | |
| yield output_seq, output_pdb, display_pdb(output_pdb), get_plddt_plot(plddt_data, S.max_t) | |
| output_seq, output_pdb, plddt = S.get_outputs() | |
| yield output_seq, output_pdb, display_pdb(output_pdb), get_plddt_plot(plddt_data, S.max_t) | |
| def get_plddt_plot(plddt_data, max_t): | |
| x = [i+1 for i in range(len(plddt_data))] | |
| fig, ax = plt.subplots(figsize=(15,6)) | |
| ax.plot(x,plddt_data,color='#661dbf', linewidth=3,marker='o') | |
| ax.set_xticks([i+1 for i in range(max_t)]) | |
| ax.set_yticks([(i+1)/10 for i in range(10)]) | |
| ax.set_ylim([0,1]) | |
| ax.set_ylabel('model confidence (plddt)') | |
| ax.set_xlabel('diffusion steps (t)') | |
| return fig | |
| def display_pdb(path_to_pdb): | |
| ''' | |
| #function to display pdb in py3dmol | |
| ''' | |
| pdb = open(path_to_pdb, "r").read() | |
| view = py3Dmol.view(width=500, height=500) | |
| view.addModel(pdb, "pdb") | |
| view.setStyle({'model': -1}, {"cartoon": {'colorscheme':{'prop':'b','gradient':'roygb','min':0,'max':1}}})#'linear', 'min': 0, 'max': 1, 'colors': ["#ff9ef0","#a903fc",]}}}) | |
| view.zoomTo() | |
| output = view._make_html().replace("'", '"') | |
| print(view._make_html()) | |
| x = f"""<!DOCTYPE html><html></center> {output} </center></html>""" # do not use ' in this input | |
| return f"""<iframe height="500px" width="100%" name="result" allow="midi; geolocation; microphone; camera; | |
| display-capture; encrypted-media;" sandbox="allow-modals allow-forms | |
| allow-scripts allow-same-origin allow-popups | |
| allow-top-navigation-by-user-activation allow-downloads" allowfullscreen="" | |
| allowpaymentrequest="" frameborder="0" srcdoc='{x}'></iframe>""" | |
| ''' | |
| return f"""<iframe style="width: 100%; height:700px" name="result" allow="midi; geolocation; microphone; camera; | |
| display-capture; encrypted-media;" sandbox="allow-modals allow-forms | |
| allow-scripts allow-same-origin allow-popups | |
| allow-top-navigation-by-user-activation allow-downloads" allowfullscreen="" | |
| allowpaymentrequest="" frameborder="0" srcdoc='{x}'></iframe>""" | |
| ''' | |
| def toggle_seq_input(choice): | |
| if choice == "protein length": | |
| return gr.update(visible=True, value=None), gr.update(visible=False, value=None) | |
| elif choice == "custom sequence": | |
| return gr.update(visible=False, value=None), gr.update(visible=True, value=None) | |
| def toggle_secondary_structure(choice): | |
| if choice == "sliders": | |
| return gr.update(visible=True, value=None),gr.update(visible=True, value=None),gr.update(visible=True, value=None),gr.update(visible=False, value=None) | |
| elif choice == "explicit": | |
| return gr.update(visible=False, value=None),gr.update(visible=False, value=None),gr.update(visible=False, value=None),gr.update(visible=True, value=None) | |
| # Define the Gradio interface | |
| with gr.Blocks(theme='ParityError/Interstellar') as demo: | |
| gr.Markdown(f"""# Protein Generation via Diffusion in Sequence Space""") | |
| with gr.Row(): | |
| with gr.Column(min_width=500): | |
| gr.Markdown(f""" | |
| ## How does it work?\n | |
| --- [PREPRINT](https://biorxiv.org/content/10.1101/2023.05.08.539766v1) --- | |
| Protein sequence and structure co-generation is a long outstanding problem in the field of protein design. By implementing [ddpm](https://arxiv.org/abs/2006.11239) style diffusion over protein seqeuence space we generate protein sequence and structure pairs. Starting with [RoseTTAFold](https://www.science.org/doi/10.1126/science.abj8754), a protein structure prediction network, we finetuned it to predict sequence and structure given a partially noised sequence. By applying losses to both the predicted sequence and structure the model is forced to generate meaningful pairs. Diffusing in sequence space makes it easy to implement potentials to guide the diffusive process toward particular amino acid composition, net charge, and more! Furthermore, you can sample proteins from a family of sequences or even train a small sequence to function classifier to guide generation toward desired sequences. | |
| 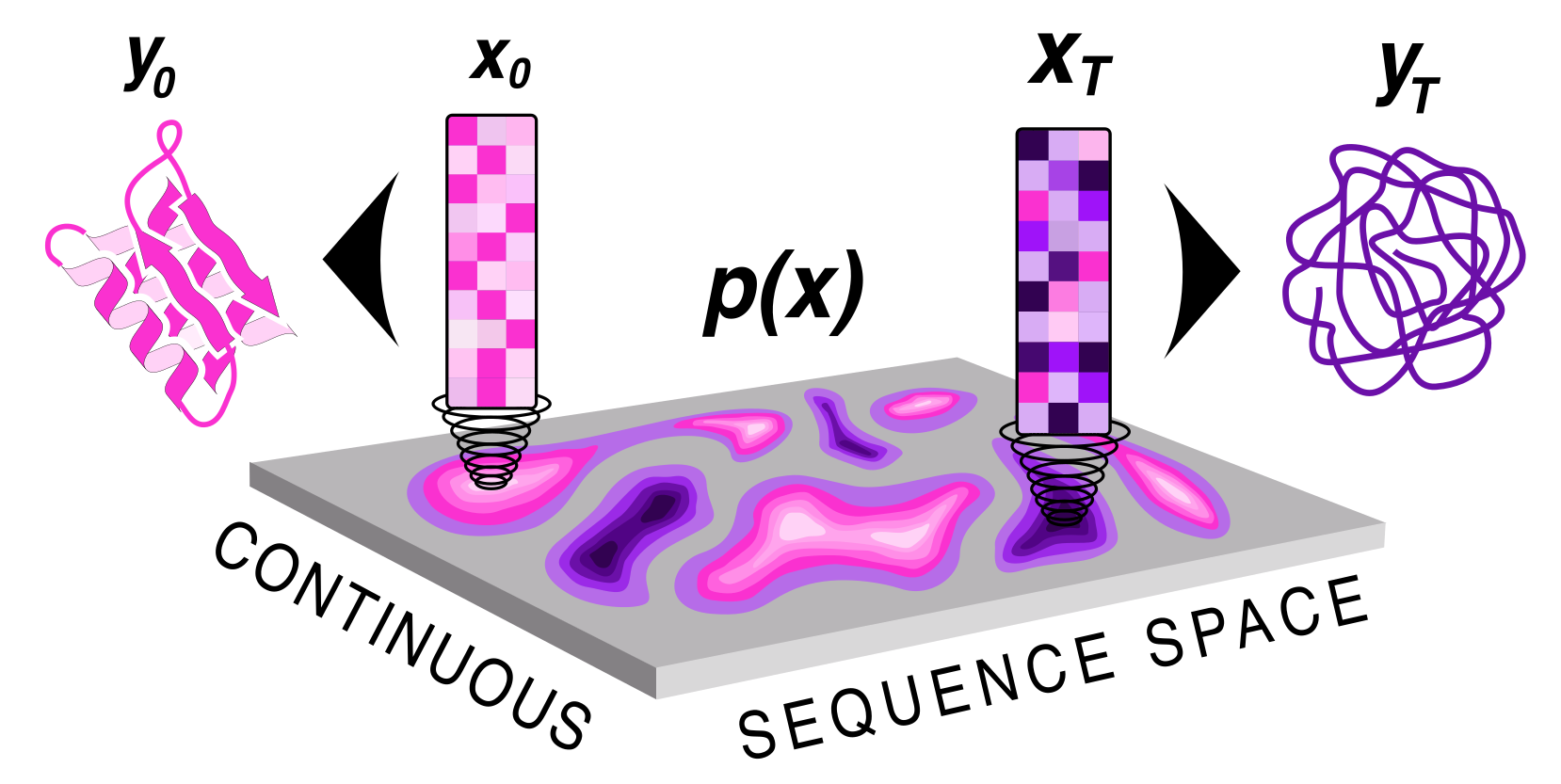 | |
| ## How to use it?\n | |
| A user can either design a custom input sequence to diffuse from or specify a length below. To scaffold a sequence use the following format where X represent residues to diffuse: XXXXXXXXSCIENCESCIENCEXXXXXXXXXXXXXXXXXXX. You can even design a protein with your name XXXXXXXXXXXXNAMEHEREXXXXXXXXXXXXX! | |
| ### Acknowledgements\n | |
| Thank you to Simon Dürr and the Hugging Face team for setting us up with a community GPU grant! | |
| """) | |
| gr.Markdown(""" | |
| ## Model in Action | |
|  | |
| """) | |
| with gr.Row().style(equal_height=False): | |
| with gr.Column(): | |
| gr.Markdown("""## INPUTS""") | |
| gr.Markdown("""#### Start Sequence | |
| Specify the protein length for complete unconditional generation, or scaffold a motif (or your name) using the custom sequence input""") | |
| seq_opt = gr.Radio(["protein length","custom sequence"], label="How would you like to specify the starting sequence?", value='protein length') | |
| sequence = gr.Textbox(label="custom sequence", lines=1, placeholder='AMINO ACIDS: A,C,D,E,F,G,H,I,K,L,M,N,P,Q,R,S,T,V,W,Y\n MASK TOKEN: X', visible=False) | |
| seq_len = gr.Slider(minimum=5.0, maximum=250.0, label="protein length", value=100, visible=True) | |
| seq_opt.change(fn=toggle_seq_input, | |
| inputs=[seq_opt], | |
| outputs=[seq_len, sequence], | |
| queue=False) | |
| gr.Markdown("""### Optional Parameters""") | |
| with gr.Accordion(label='Secondary Structure',open=True): | |
| gr.Markdown("""Try changing the sliders or inputing explicit secondary structure conditioning for each residue""") | |
| sec_str_opt = gr.Radio(["sliders","explicit"], label="How would you like to specify secondary structure?", value='sliders') | |
| secondary_structure = gr.Textbox(label="secondary structure", lines=1, placeholder='HELIX = H STRAND = S LOOP = L MASK = X(must be the same length as input sequence)', visible=False) | |
| with gr.Column(): | |
| helix_bias = gr.Slider(minimum=0.0, maximum=0.05, label="helix bias", visible=True) | |
| strand_bias = gr.Slider(minimum=0.0, maximum=0.05, label="strand bias", visible=True) | |
| loop_bias = gr.Slider(minimum=0.0, maximum=0.20, label="loop bias", visible=True) | |
| sec_str_opt.change(fn=toggle_secondary_structure, | |
| inputs=[sec_str_opt], | |
| outputs=[helix_bias,strand_bias,loop_bias,secondary_structure], | |
| queue=False) | |
| with gr.Accordion(label='Amino Acid Compositional Bias',open=False): | |
| gr.Markdown("""Bias sequence composition for particular amino acids by specifying the one letter code followed by the fraction to bias. This can be input as a list for example: W0.2,E0.1""") | |
| with gr.Row(): | |
| aa_bias = gr.Textbox(label="aa bias", lines=1, placeholder='specify one letter AA and fraction to bias, for example W0.1 or M0.1,K0.1' ) | |
| aa_bias_potential = gr.Textbox(label="aa bias scale", lines=1, placeholder='AA Bias potential scale (recomended range 1.0-5.0)') | |
| ''' | |
| with gr.Accordion(label='Charge Bias',open=False): | |
| gr.Markdown("""Bias for a specified net charge at a particular pH using the boxes below""") | |
| with gr.Row(): | |
| target_charge = gr.Textbox(label="net charge", lines=1, placeholder='net charge to target') | |
| target_ph = gr.Textbox(label="pH", lines=1, placeholder='pH at which net charge is desired') | |
| charge_potential = gr.Textbox(label="charge potential scale", lines=1, placeholder='charge potential scale (recomended range 1.0-5.0)') | |
| ''' | |
| with gr.Accordion(label='Hydrophobic Bias',open=False): | |
| gr.Markdown("""Bias for or against hydrophobic composition, to get more soluble proteins, bias away with a negative target score (ex. -5)""") | |
| with gr.Row(): | |
| hydrophobic_target_score = gr.Textbox(label="hydrophobic score", lines=1, placeholder='hydrophobic score to target (negative score is good for solublility)') | |
| hydrophobic_potential = gr.Textbox(label="hydrophobic potential scale", lines=1, placeholder='hydrophobic potential scale (recomended range 1.0-2.0)') | |
| with gr.Accordion(label='Diffusion Params',open=False): | |
| gr.Markdown("""Increasing T to more steps can be helpful for harder design challenges, sampling from different distributions can change the sequence and structural composition""") | |
| with gr.Row(): | |
| num_steps = gr.Textbox(label="T", lines=1, placeholder='number of diffusion steps (25 or less will speed things up)') | |
| noise = gr.Dropdown(['normal','gmm2 [-1,1]','gmm3 [-1,0,1]'], label='noise type', value='normal') | |
| btn = gr.Button("GENERATE") | |
| #with gr.Row(): | |
| with gr.Column(): | |
| gr.Markdown("""## OUTPUTS""") | |
| gr.Markdown("""#### Confidence score for generated structure at each timestep""") | |
| plddt_plot = gr.Plot(label='plddt at step t') | |
| gr.Markdown("""#### Output protein sequnece""") | |
| output_seq = gr.Textbox(label="sequence") | |
| gr.Markdown("""#### Download PDB file""") | |
| output_pdb = gr.File(label="PDB file") | |
| gr.Markdown("""#### Structure viewer""") | |
| output_viewer = gr.HTML() | |
| gr.Markdown("""### Don't know where to get started? Click on an example below to try it out!""") | |
| gr.Examples( | |
| [["","125",0.0,0.0,0.2,"","","","20","normal",'',''], | |
| ["","100",0.0,0.0,0.0,"","W0.2","2","20","normal",'',''], | |
| ["","100",0.0,0.0,0.0,"XXHHHHHHHHHXXXXXXXHHHHHHHHHXXXXXXXHHHHHHHHXXXXSSSSSSSSSSSXXXXXXXXSSSSSSSSSSSSXXXXXXXSSSSSSSSSXXXXXXX","","","25","normal",'',''], | |
| ["XXXXXXXXXXXXXXXXXXXXXXXXXIPDXXXXXXXXXXXXXXXXXXXXXXPEPSEQXXXXXXXXXXXXXXXXXXXXXXXXXXIPDXXXXXXXXXXXXXXXXXXX","",0.0,0.0,0.0,"","","","25","normal",'','']], | |
| inputs=[sequence, | |
| seq_len, | |
| helix_bias, | |
| strand_bias, | |
| loop_bias, | |
| secondary_structure, | |
| aa_bias, | |
| aa_bias_potential, | |
| #target_charge, | |
| #target_ph, | |
| #charge_potential, | |
| num_steps, | |
| noise, | |
| hydrophobic_target_score, | |
| hydrophobic_potential], | |
| outputs=[output_seq, | |
| output_pdb, | |
| output_viewer, | |
| plddt_plot], | |
| fn=protein_diffusion_model, | |
| ) | |
| btn.click(protein_diffusion_model, | |
| [sequence, | |
| seq_len, | |
| helix_bias, | |
| strand_bias, | |
| loop_bias, | |
| secondary_structure, | |
| aa_bias, | |
| aa_bias_potential, | |
| #target_charge, | |
| #target_ph, | |
| #charge_potential, | |
| num_steps, | |
| noise, | |
| hydrophobic_target_score, | |
| hydrophobic_potential], | |
| [output_seq, | |
| output_pdb, | |
| output_viewer, | |
| plddt_plot]) | |
| demo.queue() | |
| demo.launch(debug=True) | |